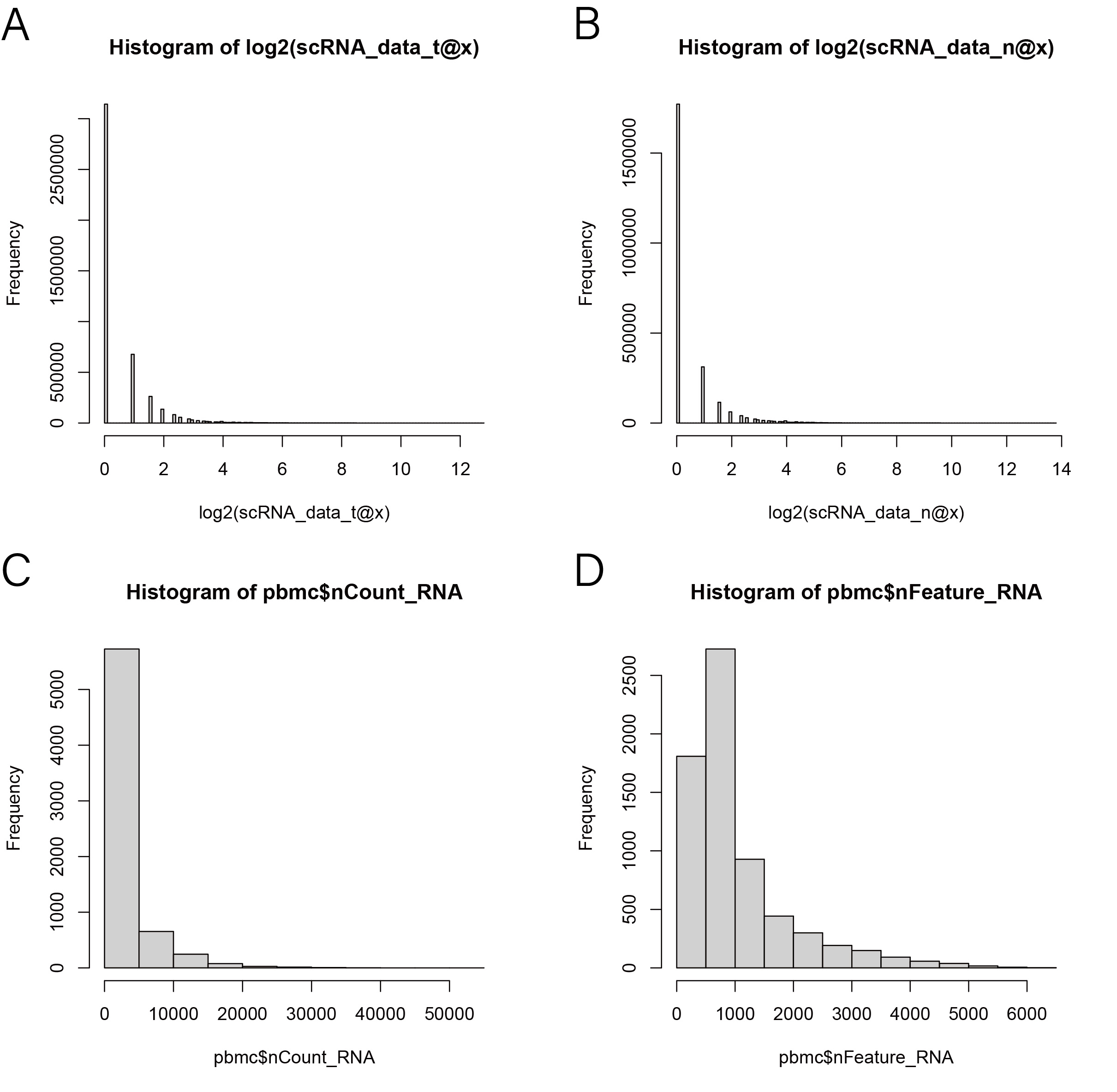
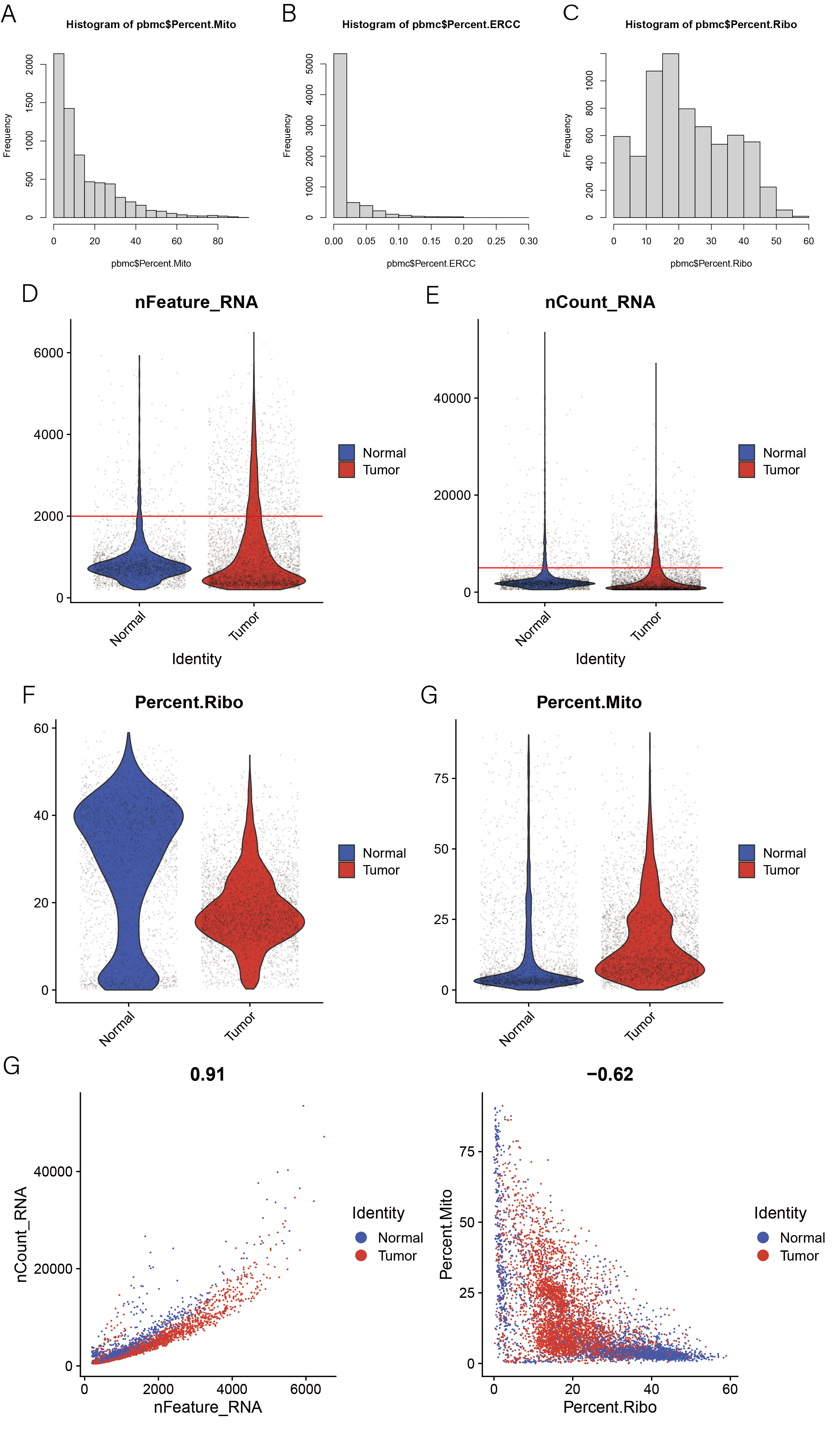
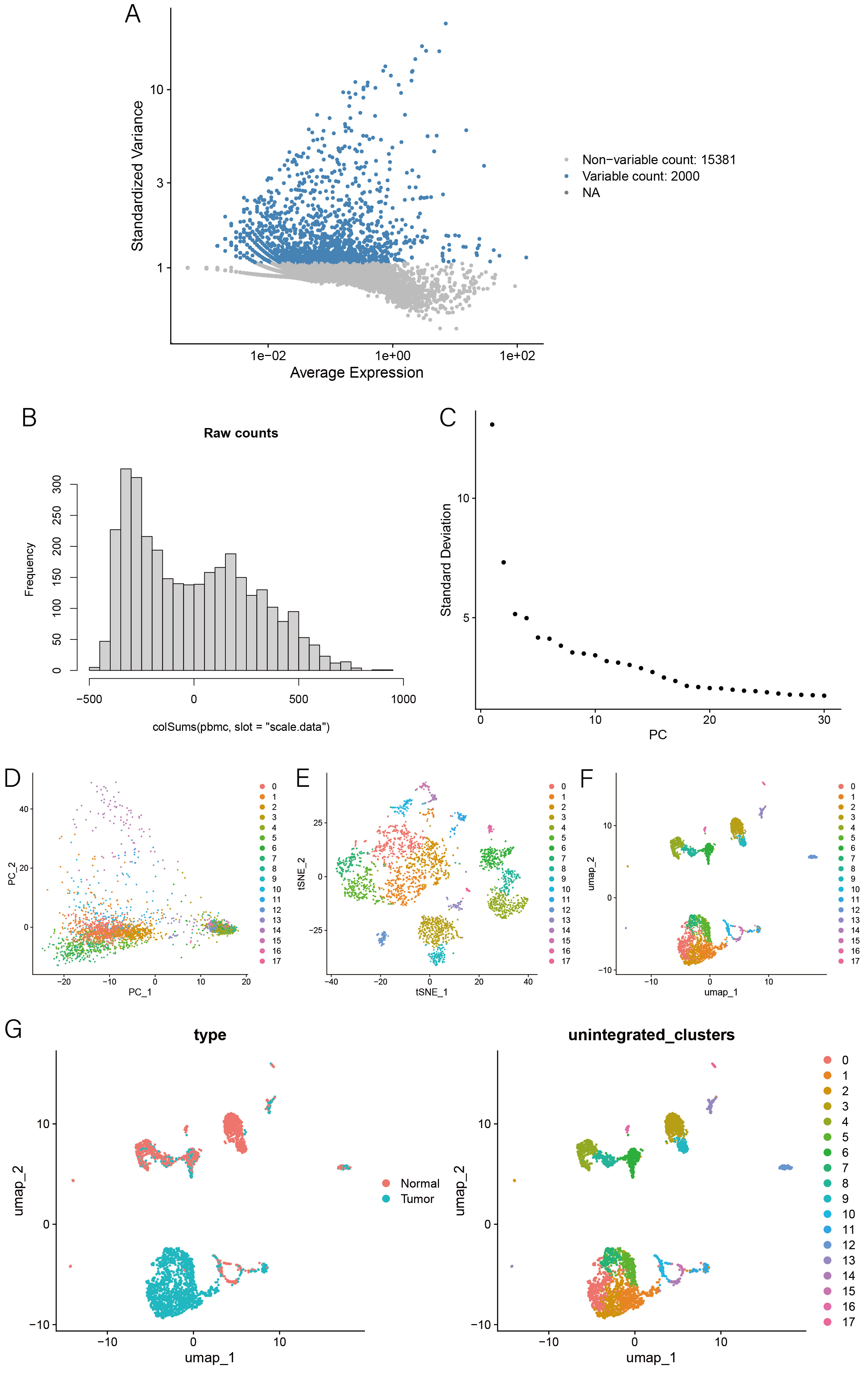
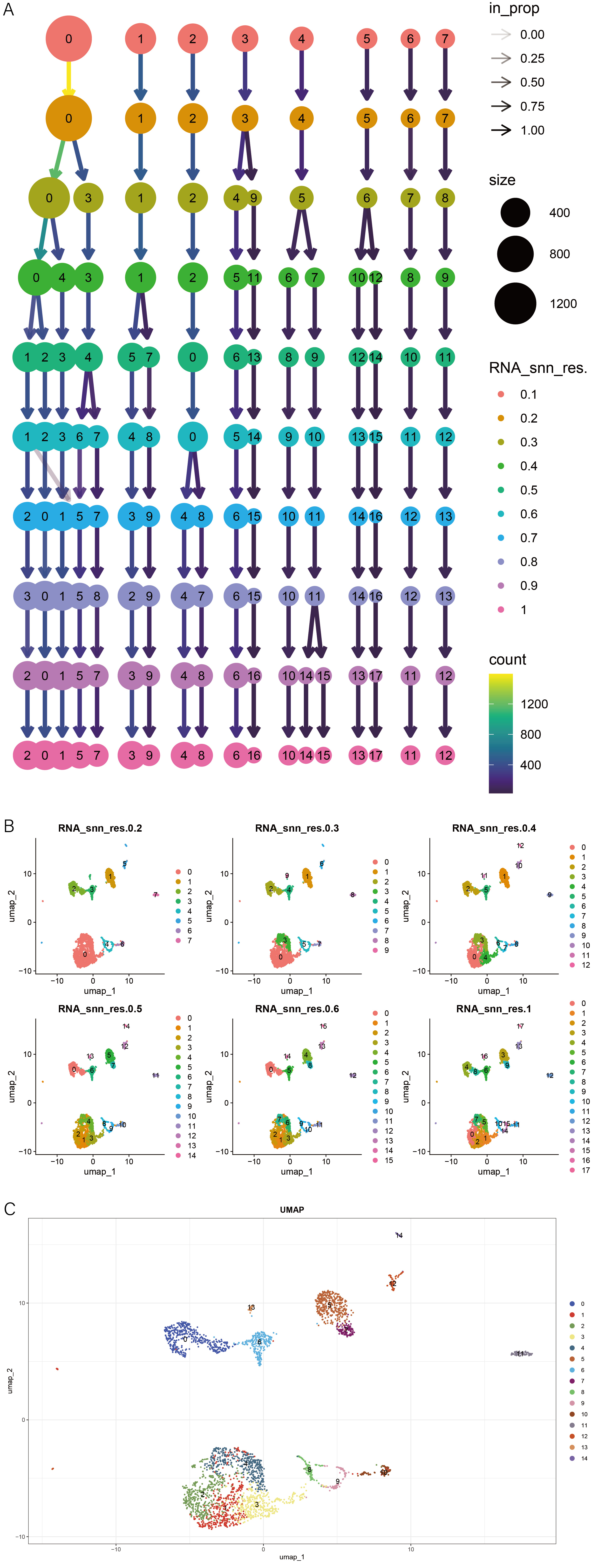
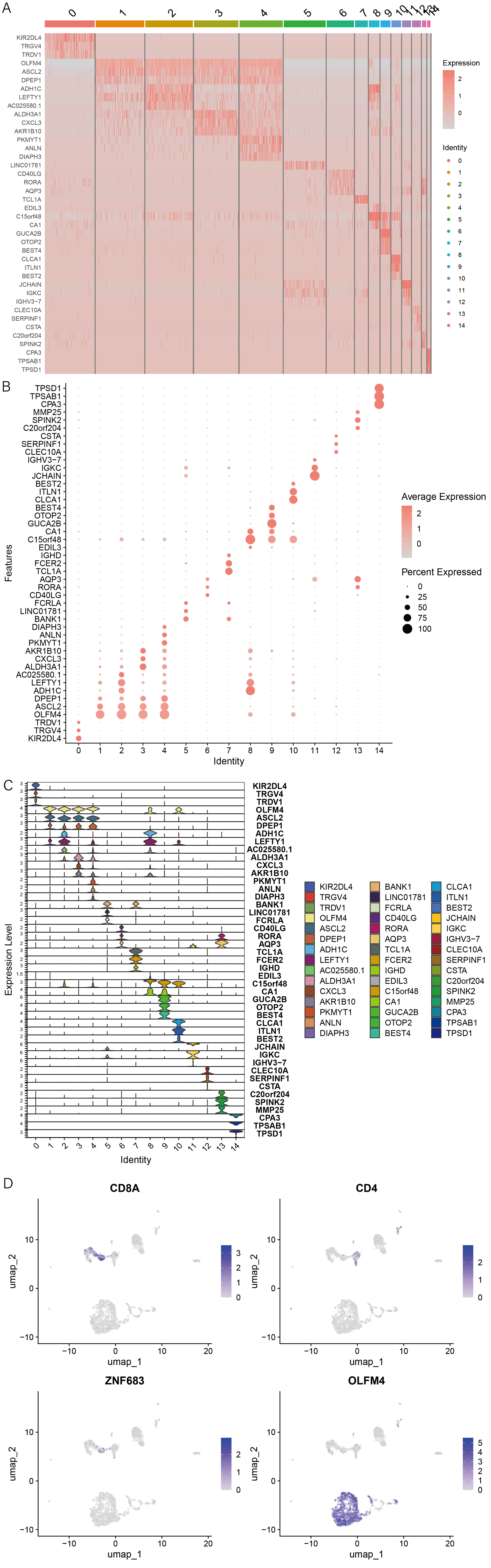
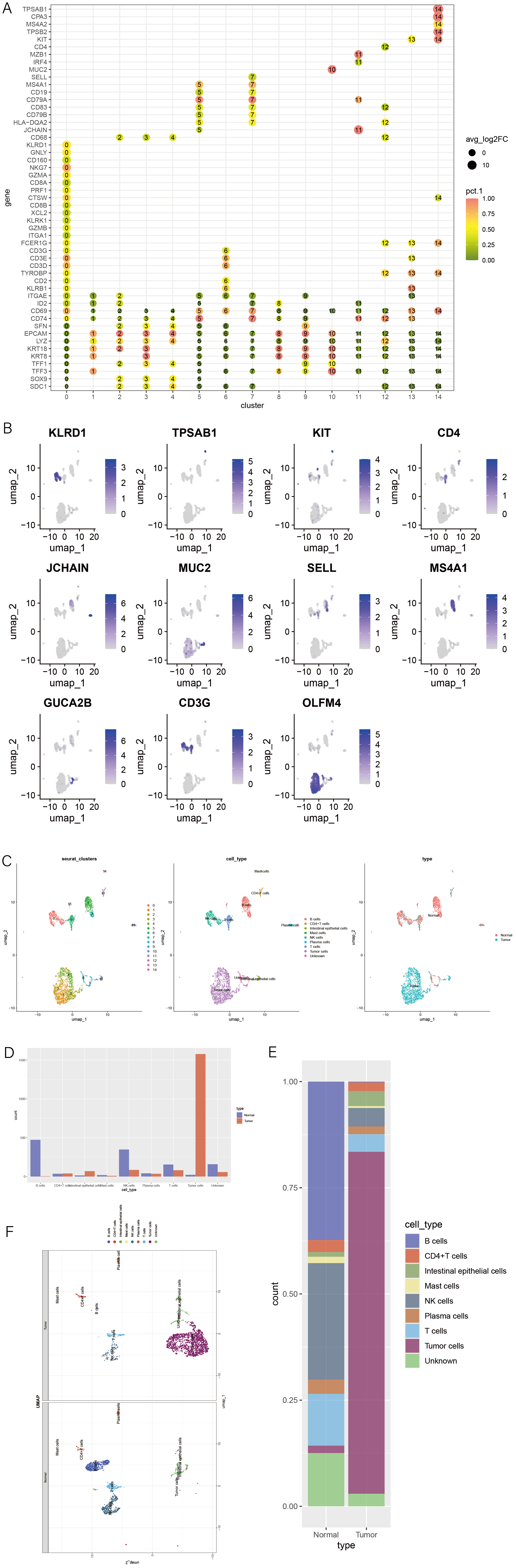
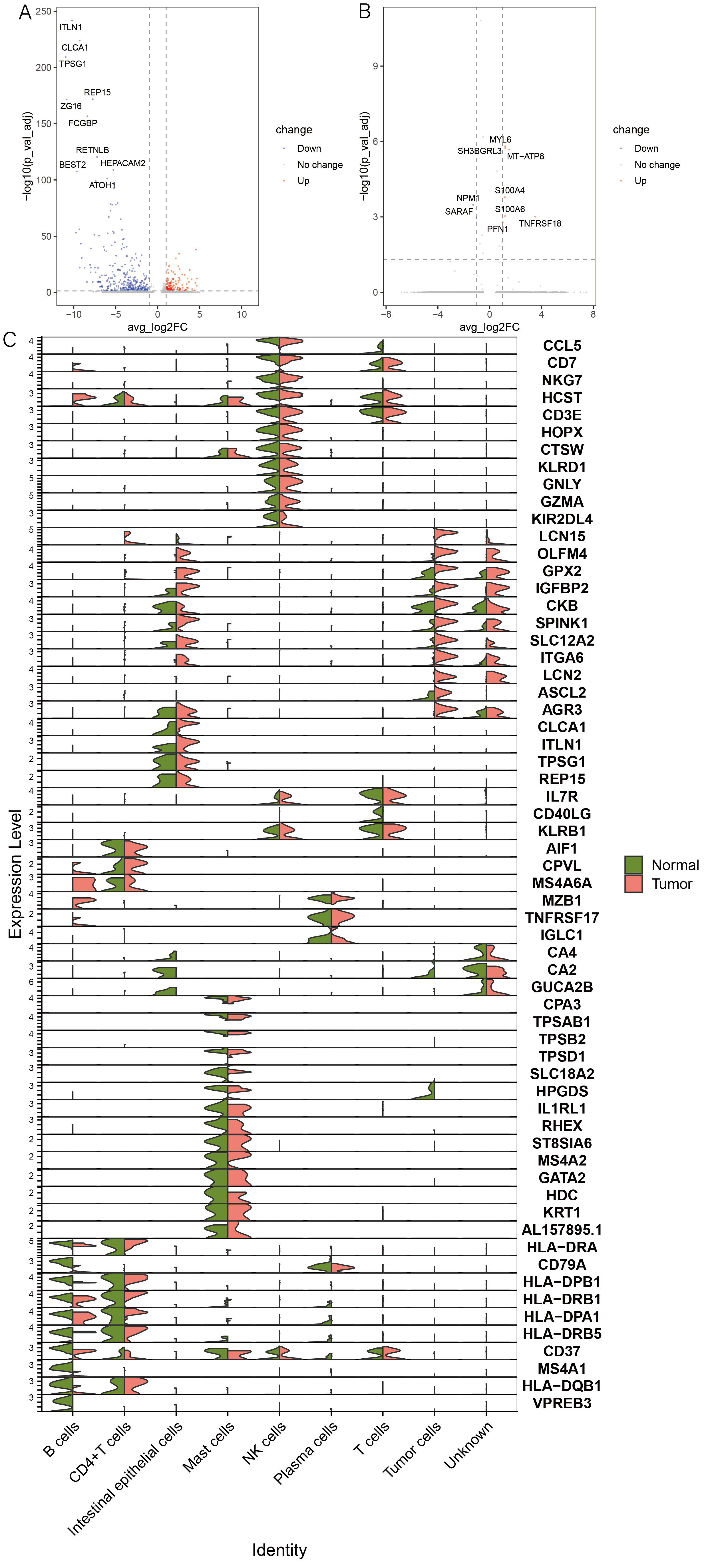
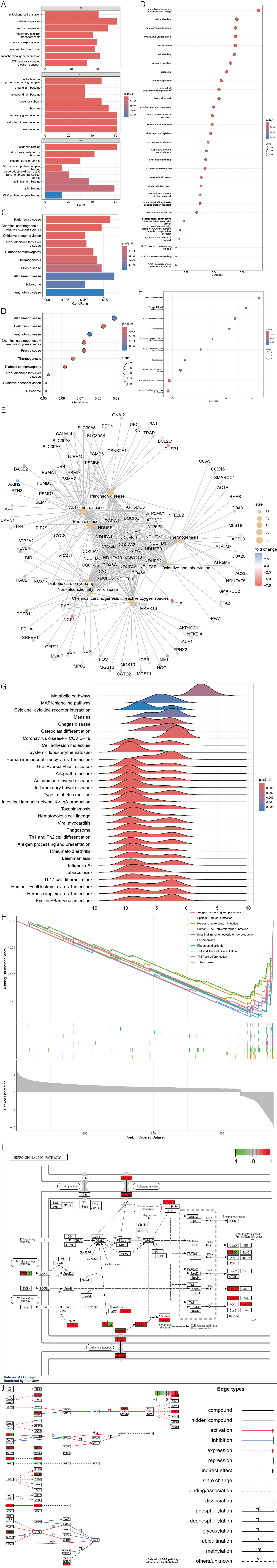
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
|
if (!require(CellChat)){
devtools::install_github("jinworks/CellChat")
devtools::install_github('immunogenomics/presto')
}
library(CellChat)
library(patchwork)
data.input <- pbmc_2[["RNA"]]$data #提取表达矩阵
labels <- Idents(pbmc_2) #提取主要分类标签,这里是cell_type
meta <- data.frame(labels = labels, row.names = names(labels))
cellchat <- createCellChat(object = pbmc_2, group.by = "ident", assay = "RNA") #构建cellchat对象
CellChatDB <- CellChatDB.human #选择人类数据库
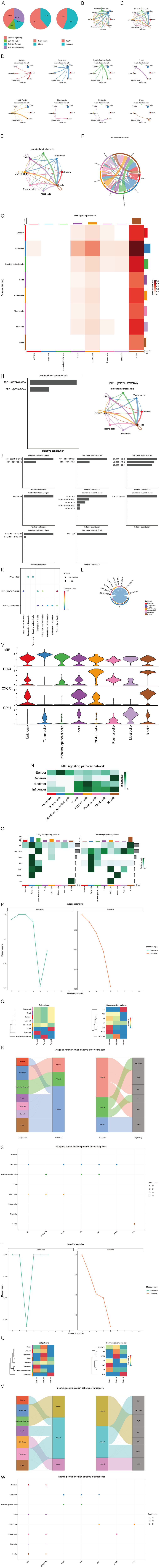
#展示该数据库中的数据类别的构成比,fig.9.A
showDatabaseCategory(CellChatDB)
ggsave(filename='DBcategory.pdf',width = 6, height = 4)
#提取数据库的子集,选择“Sevreted Signaling”部分
CellChatDB.use <- subsetDB(CellChatDB, search = "Secreted Signaling", key = "annotation")
cellchat@DB <- CellChatDB.use #将数据库更新到cellchat对象中
cellchat <- subsetData(cellchat) #从输入数据中提取与信号通路相关的配体和受体基因,不能省略
cellchat <- identifyOverExpressedGenes(cellchat) #识别在各群体中过表达的基因
cellchat <- identifyOverExpressedInteractions(cellchat) #根据过表达基因推断潜在的细胞相互作用
#使用人类蛋白质相互作用(PPI)网络平滑信号数据,改进噪声模型
cellchat <- smoothData(cellchat, adj = PPI.human) #cellchat包作者把projectData函数改为了smoothData函数!
cellchat <- computeCommunProb(cellchat, type = "triMean") #计算细胞间的通信概率,使用三均值法作为估算方式
cellchat <- filterCommunication(cellchat, min.cells = 10) #筛选通信事件,保留至少在 10 个细胞中检测到的通信对
cellchat <- computeCommunProbPathway(cellchat) #计算细胞间通讯的路径概率
cellchat <- aggregateNet(cellchat)#将细胞间通讯的网络聚合到更高的层次
groupSize <- as.numeric(table(cellchat@idents))#计算每种细胞类型的细胞数
#绘制弦图,fig.9.B
pdf(file="Interactions_string.pdf",width = 5,height = 5)
netVisual_circle(
cellchat@net$count,
vertex.weight = groupSize,
weight.scale = T,
label.edge= F,
title.name = "Number of interactions"
)
dev.off()
#绘制弦图,线的粗细为weights/strength,fig.9.C
pdf(file="Interactions_string_W_S.pdf",width = 5,height = 5)
netVisual_circle(
cellchat@net$weight,
vertex.weight = groupSize,
weight.scale = T,
label.edge= F,
title.name = "Interaction weights/strength"
)
dev.off()
#绘制每一个类型的细胞与所有的细胞之间的通讯情况,fig.9.D
mat <- cellchat@net$weight
pdf(file='plot_all.pdf',12,6)
par(mfrow = c(2,4), xpd=TRUE)
for (i in 1:nrow(mat)) {
mat2 <- matrix(0, nrow = nrow(mat), ncol = ncol(mat), dimnames = dimnames(mat))
mat2[i, ] <- mat[i, ]
netVisual_circle(
mat2,
vertex.weight = groupSize,
weight.scale = T,
edge.weight.max = max(mat),
title.name = rownames(mat)[i]
)
}
dev.off()
#单独显示一条通路,这里选择MIF通路。绘制弦图,fig.9.E
cellchat@netP$pathways#涉及到的所有的pathway
pathways.show <- c("MIF")
pdf(file = 'MIF signaling pathway network2.pdf',width = 5, height = 5)
netVisual_aggregate(
cellchat,
signaling = pathways.show,
)
dev.off()
#绘制另一种弦图,fig.9.F
pdf(file = 'chord_plot.pdf',width = 10,height = 10)
netVisual_aggregate(
cellchat,
signaling = pathways.show,
layout = "chord"
)
dev.off()
#绘制热图,fig.9.G
pdf(file = 'heatmap_plot.pdf',width = 10,height = 10)
netVisual_heatmap(
cellchat,
signaling = pathways.show,
color.heatmap = "Reds"
)
dev.off()
#计算该通路(MIF)中一些亚型的贡献占比,fig.9.H
pdf(file = "MIF_Sub.pdf",width = 5,height = 5)
netAnalysis_contribution(cellchat, signaling = pathways.show)
dev.off()
#提取MIF通路中所有亚型
pairLR.CXCR4 <- extractEnrichedLR(
cellchat,
signaling = pathways.show,
geneLR.return = FALSE
)
#提取MIF-(CD74+CRCX4)通路,画出某一条通路的弦图,fig.9.I
LR.show <- pairLR.CXCR4[1,] # show one ligand-receptor pair
pdf(file = 'MIF-CD74+CRCX4_signaling_pathway.pdf',width = 5,height = 5)
netVisual_individual(cellchat, signaling = pathways.show, pairLR.use = LR.show)
dev.off()
#可视化所有通路的亚型占比,fig.9.J
pathways.show.all <- cellchat@netP$pathways
vertex.receiver = seq(1,4)
for (i in 1:length(pathways.show.all)) {
# Visualize communication network associated with both signaling pathway and individual L-R pairs
netVisual(cellchat, signaling = pathways.show.all[i], vertex.receiver = vertex.receiver, layout = "hierarchy")
# Compute and visualize the contribution of each ligand-receptor pair to the overall signaling pathway
if (i==1){
gg <- netAnalysis_contribution(cellchat, signaling = pathways.show.all[i])
}
gg <- gg+netAnalysis_contribution(cellchat, signaling = pathways.show.all[i])
#ggsave(filename=paste0(pathways.show.all[i], "_L-R_contribution.pdf"), plot=gg, width = 3, height = 2, units = 'in', dpi = 300)
}
ggsave(file = 'contribution_of_each_pathway.pdf',width = 14,height = 10)
#绘制气泡图,本质与弦图类似,fig.9.K
pdf(file = 'bubble2.pdf',width = 5,height = 5)
netVisual_bubble(
cellchat,
sources.use = c(2), #指定信号来源的细胞群体
targets.use = c(1:8), #指定信号接收的细胞群体
signaling = c("MIF","CypA"), #指定可视化哪些信号通路
remove.isolate = FALSE #不移除信号强度低的通信对
)
dev.off()
#指定源细胞群和目标细胞群,以及指定信号通路,绘制弦图,fig.9.L
pdf(file = 'Tumor_to_all_chord.pdf',width = 7,height = 5)
netVisual_chord_gene(
cellchat,
sources.use = c(2), #指定信号来源的细胞群体,编号为 1 到 8。
targets.use = c(1:8), #指定信号接收的细胞群体,编号为 1 到 8。
signaling = c("MIF"),
legend.pos.x = 8 #调整图在画布上的位置
)
dev.off()
#绘制与指定信号通路相关的基因的表达的小提琴图,fig.9.M
pdf(file = 'cell_chat_Vlnplot_MIF.pdf',width = 7,height = 5)
plotGeneExpression(
cellchat,
signaling = "MIF", #指定一条信号通路
enriched.only = TRUE, #只显示显著上调的基因
type = "violin" #绘制小提琴图
)
dev.off()
#计算信号网络中各细胞群体的中心性指标(如度数、介数和接近中心性等)
cellchat <- netAnalysis_computeCentrality(
cellchat,
slot.name = "netP" #指定数据来源为概率加权信号网络netP
)
#绘制图像,可视化在该通路下每个细胞的作用如何,分为Sender、Receiver、Mediator和Influencer,fig.9.N
pdf(file = 'SignalingRole_heatmap.pdf',width = 7,height = 5)
netAnalysis_signalingRole_network(
cellchat,
signaling = pathways.show, #指定绘图的信号通路,这里是MIF
width = 8,
height = 2.5,
font.size = 10
)
dev.off()
#生成热图,展示每个细胞群体在信号网络中作为发送者(Sender)和接收者(Receiver)的能力,fig.9.O
ht1 <- netAnalysis_signalingRole_heatmap(
cellchat,
pattern = "outgoing"
)
ht2 <- netAnalysis_signalingRole_heatmap(
cellchat,
pattern = "incoming"
)
pdf(file = 'O_R_heatmap.pdf',width = 12,height = 7)
ht1+ht2
dev.off()
library(NMF)
library(ggalluvial)
###outgoing###
#有两个参数:
#Cophenetic相关系数,用于衡量数据在聚类时的保真度(fidelity)
#Silhouette系数衡量每个点在聚类中的紧密度和聚类之间的分离度
#fig.9.P
K <- selectK(cellchat, pattern = "outgoing")
pdf(file = 'K_out.pdf',width = 12,height = 7)
K
dev.off()
#绘制聚类热图,fig.9.Q
pdf(file = 'CommuPattern_out.pdf',width = 12,height = 5)
nPatterns = 3
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "outgoing", k = nPatterns)
dev.off()
#聚类-riverplot-out,fig.9.R
pdf(file = 'riverplot_out.pdf',width = 12,height = 7)
netAnalysis_river(cellchat, pattern = "outgoing")
dev.off()
#聚类-dotplot-out,fig.9.S
pdf(file = 'dotplot_out.pdf',width = 12,height = 7)
netAnalysis_dot(cellchat, pattern = "outgoing")
dev.off()
###incoming###
#fig.9.T
K <- selectK(cellchat, pattern = "incoming")
pdf(file = 'K_in.pdf',width = 12,height = 7)
K
dev.off()
#聚类热图,fig.9.U
pdf(file = 'CommuPattern_in.pdf',width = 12,height = 5)
nPatterns = 3
cellchat <- identifyCommunicationPatterns(cellchat, pattern = "incoming", k = nPatterns)
dev.off()
#聚类-riverplot-in,fig.9.V
pdf(file = 'riverplot_in.pdf',width = 12,height = 7)
netAnalysis_river(cellchat, pattern = "incoming")
dev.off()
#聚类-dotplot-in,fig.9.W
pdf(file = 'dotplot_in.pdf',width = 12,height = 7)
netAnalysis_dot(cellchat, pattern = "incoming")
dev.off()
|