Bulk-RNA测序上游分析——从reads到counts
转录组基础知识
以下记载了转录组最基础的一些知识,这些概念在RNA测序分析中是至关重要的:
1、基因组Genome:指某一特定物种细胞内或病毒颗粒内的一整套遗传物质(DNA或RNA)。对于人类而言,人类细胞是二倍体,有2套染色体,其中任何一套染色体的DNA序列即为一个基因组,比如人类基因组就是1-22号染色体+X+Y。在广义上,线粒体DNA也可以算作基因组的一部分。
2、转录组Transcriptome:广义上指在某种条件下在一个细胞或一群细胞中所转录出的RNA总和,包括mRNA、rRNA、tRNA和其他非编码RNA等。狭义的转录组特指转录出的mRNA。转录组和基因组的差别在于,基因组在给定的细胞株上一般不变,而转录组可能会有很大变化,它反映了在给定条件下细胞内活跃表达的基因,故也成为“基因表达谱”。
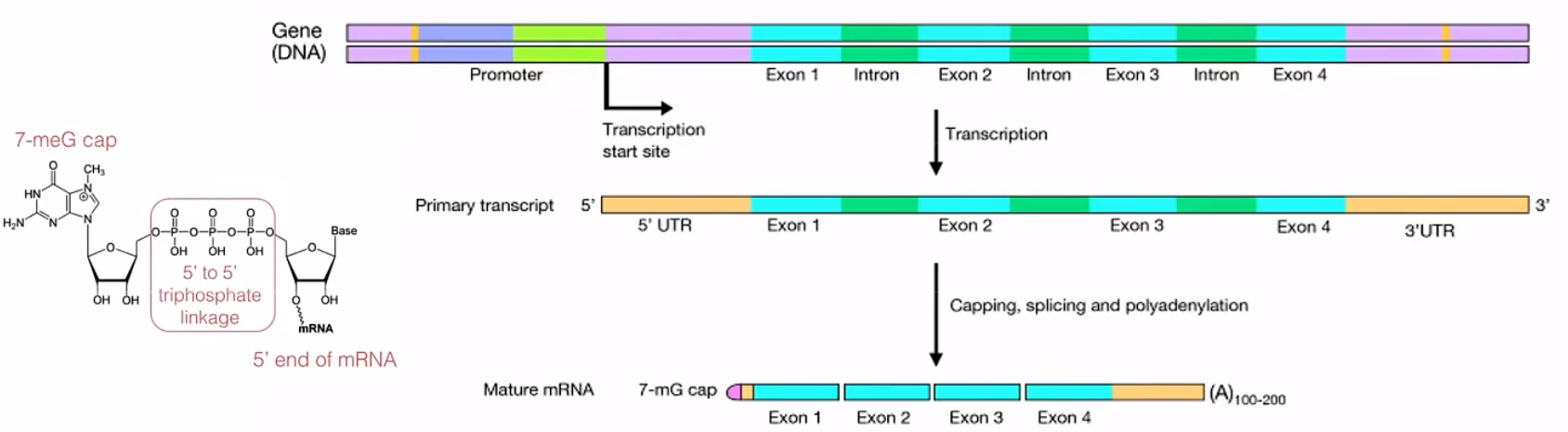
3、转录Transcription:即在RNA聚合酶的作用下,遗传信息由DNA复制到RNA的过程。其分为三个过程:启动、延伸和终止。由RNA聚合酶和转录因子共同结合到模板DNA的启动子序列上开启转录过程,接着按互补配对原则进行延伸,最后通过依赖ρ因子的终止或非依赖ρ因子的终止来终止转录。转录出的初级转录RNA包括以下区域:5’UTR(5’非翻译区)、3’UTR(3’非翻译区)、Intron(内含子)、Exon(外显子)。
4、转录后修饰Post-transcriptional modification:是指在真核细胞中,将初级转录RNA转化为成熟RNA的过程。包括3个过程:5’端加帽、3’端加尾以及RNA剪切。
5、可变剪接Alternative splicing:又称可变剪接,即一条未经剪接的RNA,含有的多种外显子被剪成不同的组合,因而翻译成不同的蛋白质。剪切出的不同mRNA即为mRNA异构体(isomer)。
6、转录本Transcript:即一条mRNA。一条基因可能有不同的转录本,其中一个原因就是可变剪切。
硬件和软件的准备
1、个人计算机的准备:
-
操作系统:Ubuntu 24.04 LTS
-
RAM:16G;硬盘:500G
2、软件的准备。
-
不具体介绍某个软件的下载方法
-
一般的软件安装命令方法如下:
1sudo apt install Name_of_Software -
或者直接在Github等网站上下载
3、将软件所在位置添加入环境变量,或将PYTHON脚本添加到PYTHON环境变量:
|
|
FASTQ和FASTA文件概述
RNA测序分析的第一步是从测序的原始数据开始的。测序公司返回的原始数据文件是FASTQ格式,里面的每一条记录就是一个read读段。还有一种类似的数据格式叫做FASTA,一般用来记录已经组装好的、参考基因组的序列。
使用以下命令下载本例所需的文件:
|
|
其中,前两个文件为来自淋巴母细胞系的双端测序文件,数据在加利福尼亚理工学院产生,来自早期Illumina平台。下载完毕后解压并重命名为Read1.fastq,Read2.fastq。第三个文件是从ENSRMBL网站上下载的人类参考基因组,解压后大小约为30Gb。下载完解压并重命名为Homo_sapiens.GRCh37.74.dna.toplevel.fa。
分别打开Read1.fastq和Read2.fastq两个文件,可以发现其前四行分别为:
|
|
以及
|
|
Read1.fastq和Read2.fastq中每一条记录都是一一对应的,代表同一条mRNA的两端的序列。每一条fastq记录都由4行组成:
-
第一行记录了测序时的相关信息,其中@HWUSI-EAS1665_28是测序机器名称,5代表测序槽lane的序号,1代表测序小室tile的编号,1038和939分别是tile内的XY坐标,/1代表是正向测序序列,/2代表是逆向测序序列。
-
第二行是测得的mRNA序列,本里中所有的记录都是75bp的,用ATCG代表四种碱基,N代表不确定的模糊碱基。
-
第三行是一个加号(+)作为分隔符。
-
第四行是碱基的质量Q,Q为-10*lg(P)再四舍五入到整数,其中P是碱基错误的概率,再将Q加上33转化为ASCII码(phred33编码)【如果是低于1.8版本的Illumina软件产生的FASTQ文件则是Q+64转化为ASCII码(phred64编码),本例就是phred64编码的】。
接着,打开Homo_sapiens.GRCh37.74.dna.toplevel.fa,其前两行为:
|
|
该文件的每条记录都由2行组成:
-
第一行记录了基因组的相关信息,本例中>1代表是1号染色体,dna:chromosome代表记录的是染色体DNA,chromosome:GRCh37代表是人类基因组版本GRCh37的数据,1代表染色体编号为1,1:249250621代表起始位置从1到249250621,1代表正链,REF说明这是个参考序列。
-
第二行即为基因序列。
质量控制与预处理
1、用fastqc对原始文件生成一个质量报告。
|
|
2、使用Trimmomatic【Trimmomatic版本0.39】过滤掉平均质量低于20(AVGQUAL:20)的read,采用双端测序模式(PE)。
|
|
其中生成的upaired1/2.fq.gz文件存储了经过过滤幸存、但失去其配对的read。
3、使用Trimmomatic修剪,当碱基质量低于20(TRAILING:20)时从3’端修剪碱基,并且去除修剪后长度低于50bp(MINLEN:50)的read。该命令作用于双端read。其他修剪方法暂略。
|
|
4、接头的去除。案例中原始数据已经进行接头的去除,这里暂略接头的去除方法。
5、使用prinseq-lite【prinseq-lite版本0.20.4】进行筛选。去除长度小于50的read(-min_len 50),去除平均质量低于20的read(-min_qual_mean 20)【这一工作在已经在1和2中完成,可以省略】;去除低复杂度的read,即熵<70的read(lc_method entrophy lc_threshold 70);去除模糊碱基N>2的read(ns_max_n 2)。经过滤过后保留配对的文件存储在nfiltered_1/2.fastq中,经过滤过幸存、但失去其配对的read保存在nfiltered_1/2_single-tons.fastq中。这一步骤可能要计算数个小时。
|
|
6、重复read的删除。在RNA-seq中,重复往往是对高度表达的转录本进行测序的自然结果(高表达导致测序重复数多),不建议删除重复。如有必要可以用prinseq-lite来处理,删除出现100次以上(-derep_min 101)的准确重复段(-derep 1)。
|
|
将read比对到参考基因组
1、如前所述,在Ensembl上下载人类参考基因组:在http://ftp.ensembl.org/pub/release-74/fasta/homo_sapiens/dna上下载Homo_sapiens.GRCh37.74.dna.toplevel.fa.gz文件并解压,解压后的文件大小约为33Gb。
|
|
2、用bowtie2【bowtie2版本2.5.4】生成索引。
|
|
该操作会创建6个bt2l文件,均以CRCh37.74作为索引基本名。该操作会花费数个小时。生成的6个文件是GRCh37.74.1.bt2l;GRCh37.74.2.bt2l;GRCh37.74.3.bt2l;GRCh37.74.4.bt2l;GRCh37.74.rev.1.bt2l;GRCh37.74.rev.2.bt2l。他们的基本名是CRCh37.74。
3、将read比对到基因组。这里参考的索引基本名是CRCh37.7(-x CRCh47.7),输入文件(-U Read1.fastq.gz)是fastq格式(-q),它使用phred+64编码(–phred64)。将结果(-S)输出到Read1aligned.sam的文件,这里面不包括未比对的read(–no-unal)。有8个处理器同时使用以加快对比速度(-p 8)。该操作会花费数个小时。
-
如果是单端测序:
1bowtie2 -q --phred64 -p 8 --no-unal -x GRCh37.74 -U Read1.fastq.gz -S Read1aligned.sam -
如果是双端测序:
1bowtie2 -q --phred64 -p 8 --no-unal -x GRCh37.74 -1 Read1.fastq.gz -2 Read2.fastq.gz -S Readaligned.sam
#Bowtie2在进行比对时不需要原始的fasta文件,只需要用生成的6个索引文件即可完成比对。比对结束后Bowtie2按照SAM格式报告比对结果,本例中文件名为Readaligned.sam。
4、使用samtools【samtools版本1.19.2】对SAM文件进一步处理。
-
将SAM格式转化为BAM格式以节省空间,并且方便后续的操作。本例中指定输入是SAM文件(-S),输出是BAM文件(-b),输出文件名是alignments.bam(-o alignments.bam)。
1samtools view -bS -o alignments.bam Readaligned.sam -
在BAM中按染色体坐标进行排序,或者使用名称排序。
1samtools sort -o alignments.sorted.bam alignments.bam -
给BAM文件编制索引。
1samtools index alignments.sorted.bam这将会生成一个BAI索引文件alignments.sorted.bam.bai
-
指定一个染色体或染色体区域,制作对比的子集。本例提取18号染色体的比对制作比对的子集。
1samtools view -b -o alignments.sorted.18.bam alignments.sorted.bam 18 -
基于作图质量过滤比对,本例中保留作图质量高于30的比对。
1samtools view -b -q 30 -o alignments_MQmin30.bam alignments.bam -
查看统计项目,如多少个read正确配对,或多少个read与其配对read作图到不同染色体等。
1samtools flagstat alignment.bam -
BAM文件索引添加“chr”,如将“1”变成“ch1”(如果需要):
1samtools view -H H1Rep1.18.bam | sed -e 's/SN:\([0-9XY]\)/SN:chr**\1**/' -e 's/SN:MT/SN:chrM/' | samtools reheader - H1Rep1.18.bam > H1Rep1.18.chradded.bam
4、可视化比对的read。可以使用Intergrative Genomics Viewer(IGV)【IGV版本2.4.10】软件对BAM文件进行可视化。可视化过程需要两个文件,即alignments.sorted.bam,以及它的索引文件alignments.sorted.bam.bai。本例中java版本需要切换到java8才能运行IGV
讨论
讨论1:为什么要制作参考基因组索引?
将一条read(本例中为75bp)匹配到数以亿计bp的人类基因组并非一件容易的事,而匹配一个样本全部数十万条记录可能会花上数百年的时间。对参考基因组进行预处理,就如同给字典加上目录一样,在查询的时候只需要从目录定位大致位置,再在小范围内查找即可(而不是遍历整个字典)。为参考基因组制作索引可以大大减小比对的时间复杂度,从而优化查找时间。Bowtie2采用的是FM-index压缩技术,该技术基于Burrows-Wheeler Transform(BWT)和Suffix Array(后缀数组)算法,能够在占用较小内存的同时实现快速的子字符串匹配。此外,制作的索引包含了原始fasta文件的所有信息,故bowtie2在进行匹配时只需要用到索引即可,不需要原始fasta文件。在本例中:
-
GRCh37.74.1.bt2l、GRCh37.74.2.bt2l包含了FM-index的信息;
-
GRCh37.74.3.bt2l、GRCh37.74.4.bt2l包含了额外的索引结构辅助快速定位;
-
GRCh37.74.rev.1.bt2l、GRCh37.74.rev.2.bt2l包含了反向互补序列的FM-index信息。
此外,这些都是二进制文件,无法直接读取。
讨论2:SAM文件格式
SAM格式储存了bowtie2将read与参考基因组进行比对后的结果。SAM文件内容如下:
|
|
其中:
-
@HD行为文件头信息,包括版本(VN)、排序方式(SO)和排序方式(GO)
-
@SQ行为参考序列信息,包括染色体名称(SN)和长度(LN)
-
@PG行为程序信息,包括程序ID(ID)、程序名(PN)、版本号(VN)和指令(CL)
-
接下来每行都记录了一条比对信息,由11个必须字段和若干可选字段组成,中间用Tab分隔:
| 字段编号 | 字段名称 | 说明 |
|---|---|---|
| 1 | QNAME | 序列名称,本例为HWUSI-EAS1665_28:5:1:1242:950 |
| 2 | FLAG | 比对标志位,用于描述比对的特征和属性,这里153转化为2进制为10011001,代表8个参数 |
| 3 | RNAME | 比对到的参考序列名称,这里1代表比对到1号染色体 |
| 4 | POS | 对比的起始位置,这里之1号染色体的76228973位 |
| 5 | MAPQ | 比对的可信度 |
| 6 | CIGAR | CIGAR字符串,这里75M指75个碱基完全匹配。不同字母所代表的含义不做展开 |
| 7 | RNEXT | 指当前read的配对read比对到的参考序列名称。“=”代表二者对比到了同一个参考序列 |
| 8 | PNEXT | 下一个比对的起始位置 |
| 9 | TLEN | 模板长度,表示该read在参考序列上的覆盖长度。这里0代表数据不可用 |
| 10 | SEQ | read序列 |
| 11 | QUAL | read质量,phred64编码 |
| +1 | AS:i | 比对得分 |
| +2 | XN:i | 模糊匹配数,即匹配到多个位置的数量 |
| +3 | XM:i | 错配数 |
| +4 | XO:i | 比对过程中发生插入/删除(indel)事件的数量 |
| +5 | XG:i | 比对中发生插入或删除碱基的总数 |
| +6 | NM:i | 编辑距离 |
| +7 | MD:Z | read和参考序列之间的错配位置与类型 |
| +8 | YT:Z | 比对类型 |
定量和基于注释的质量控制
1、在UCSC Table Browser上获得人类基因组注释文件:在assembly菜单选择Feb. 2009 (GRCh37/hg19);Group菜单选择Genes and gene predicitions;Track菜单选择Ensembl gene;region选择genome;output format选择BED – browser extensible data;output file写hg19_Ensembl_chr.bed(因为Ensembl数据集中染色体前缀包括“chr”)。
2、准备数据集
-
在ensembl上下载18号染色体的fasta文件。
1wget -c https://ftp.ensembl.org/pub/release-74/fasta/homo_sapiens/dna/Homo_sapiens.GRCh37.74.dna.chromosome.18.fa.gz -
使用wget命令下载wgEncodeCaltechRnaSeqH1hescR2x75Il200AlignsRep1V2.bam文件,它来自人类胚胎干细胞系,数据在加利福尼亚理工学院产生,来自早期Illumina平台。
1wget -c http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeCaltechRnaSeq/wgEncodeCaltechRnaSeqH1hescR2x75Il200AlignsRep1V2.bam -
下载对应的注释文件。
1wget -c http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeCaltechRnaSeq/wgEncodeCaltechRnaSeqH1hescR2x75Il200AlignsRep1V2.bam.bai为方便起见将文件分别重命名为H1Rep1.bam和H1Rep1.bam.bai
-
从H1Rep1.bam中提取比对到18号染色体的那一部分。
1samtools view -b -o H1Rep1.18.bam H1Rep1.bam chr18 -
将H1Rep1.18.bam排序后制作索引
1 2samtools sort -o H1Rep1.18.sorted.bam H1Rep1.18.bam samtools index H1Rep1.18.sorted.bam
3、使用seq命令删除hg19_Ensembl_chr.bed文件中染色体名称的chr前缀(如有必要)。
|
|
4、使用RSeQC中的read_distribution.py计算read对不同基因组特征类型的分布。
|
|
结果将以表格形式报告read和tag的总数以及所属类型。
5、使用RSeQC中的geneBody_coverage.py可以生成一个覆盖图来检查转录本的覆盖是否均匀,即是否存在3’端或5’端偏差。
|
|
运行该程序需要R在$PATH上。
6、使用RPKM_saturation.py对read的子集重新取样,对每个子集按RPKM单位计算丰度,并检查他们是否稳定。
|
|
7、使用junction_annotation.py将剪接点划分为“新的”、“部分新”的和“已知的”,分别代表0个、1个、2个剪接位点在参考基因模型中。结果以饼图显示。
|
|
8、使用junction_saturation.py检查剪接点的测序饱和状态。软件将检测到的剪接点标记为新的、部分新的和已知的,并通过重新抽样分析其饱和状态。
|
|
9、使用HTSeq-count输出每个基因的计数。HTSeq-count需要SAM/BAM格式的比对文件,以及一个GTF格式的基因组注释文件。Htseq-count期望双端数据按读段名称进行排序,同时BAM文件和注释文件应当有相同的基因名称,本例中需要将GTF文件中基因名称1、2…转化为chr1、chr2…
|
|
10、为了方便对差异进行统计检验,最好删除counts.txt的后5行。
|
|
至此,我们已经由原始的reads得到了每个基因的counts。下一步,我们将从counts往后进行分析。
讨论
讨论1:如何度量RNA-Seq数据的质量?
RNA-Seq读段基质量问题,如低置信度碱基问题,已经可以在原始读段水平上检测出,并且用phred64编码的形式报告在fastq文件中。当读段被作图到一个参考基因组,并且它们的位置与注释相匹配时,则可以进一步度量读段的质量,包括:
-
测序深度的饱和性:检查测序深度是否接近饱和。
-
不同基因组特征之间的读段分布:比如读段在外显子、内含子以及基因间区的分布情况;读段在5’UTR和3’UTR之间的分布情况;读段在蛋白质编码基因、假基因、rRNA、miRNA之间的分布情况等等。
-
沿转录本的覆盖均匀性:不同的实验方法可能引入不同的位置偏差,例如一个polyA尾捕获步骤可能导致读段主要来自转录本的3’端。
讨论2:如何度量基因表达丰度?
每个转录本生产呢个的read数目取决于很多因素,例如较长的转录本可能会产生较多的read,此外,转录组组成、GC偏差和随机引物也可能造成序列特异性偏差。为了比较不同不同基因之间的表达差异需要进行归一化处理。其中一个指标RPKM(reads per kilobase per million mapped reads,每百万作图的读段每千碱基的读段),由Mortazavi et.al引入:
$$ \text{RPKM} = \frac{\text{numReads}}{\left(\frac{\text{geneExonLength}}{1000}\right) \times \left(\frac{\text{totalNumReads}}{1000000}\right)} $$即,认为一条基因的read数与基因长度和总read数成正比,故将其作为分母除来归一化。这样可以在同一样本中比较不同基因的表达差异。
对于双端测序,可以使用指标FPKM(fragments per kilobase per million mapped reads,每百万作图的读段每千碱基的片段):
$$ \text{FPKM} = \frac{\text{numFragments}}{\left(\frac{\text{geneExonLength}}{1000}\right) \times \left(\frac{\text{totalNumFragments}}{1000000}\right)} $$这与RPKM非常的类似,Fragments指的是双端都匹配到同一个转录本上的read对,并且只计算一遍。此外,另一个指标称为TPM(transcript per million,每百万转录本):
$$ TPM_i = \frac{\frac{N_i}{L_i} \times 10^6}{\sum_{k=1}^{n} \frac{N_k}{L_k}} = \frac{RPKM_i}{\sum_{k=1}^{n} RPKM_k} \times 10^6 $$其中Ni指匹配到第i个转录本的read数,Li是第i个转录本的长度。TPM相当于是将RPKM或FPKM归一化,使得在不同样本中可以比较相同基因的表达差异。
讨论3:操作中输出的counts.txt的数据结构。
该文件的最后几行如下所示:
|
|
最后5行分别代表没有进行计数的原因和对应的读段数目:比对与任何基因均不重叠、其比对与多个基因重叠、对比质量低于阈值、完全没有比对以及比对到基因组中的多个位置。
Bulk-RNA测序下游分析——从counts开始
数据准备
|
|
使用R语言进行分析
1、将7个样本的counts合并到一个表格,然后保存在本地。在本例中,7个txt文件在工作目录下的data文件夹里。
|
|
2、用DESeq包进行差异分析,差异基因存储在sig.deseq变量中。
|
|
3、除此之外,还可以用不同的包进行分析,最后将几种方法得出的差异基因取交集。
|
|
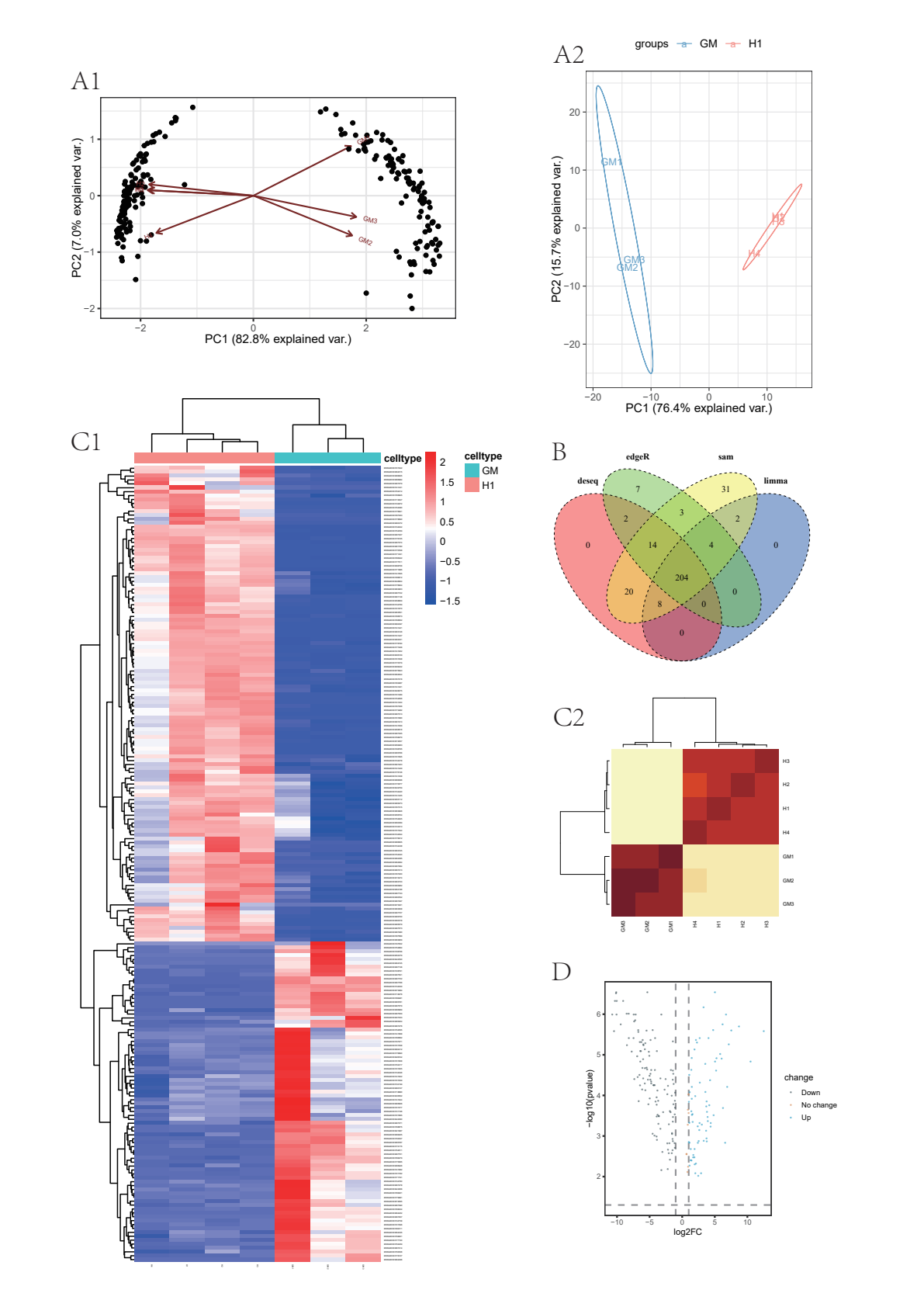
4、进行可视化处理,包括:进行主成分分析判断两组数据有何差异;对四种分析方法分析出的差异基因做Venn图并找出共有的差异基因;绘制热图可视化基因表达;绘制火山图进一步筛选基因。
-
主成分分析
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55# PCA GENE fig.A1 if (!require(ggbiplot)) install.packages("ggbiplot") library(ggbiplot) ct <- count.table[rownames(count.table) %in% sig.deseq,] ct = t(scale(t(ct))) ct.pca <- prcomp(ct,scale.=TRUE) ggbiplot( ct.pca, choices=c(1,2), obs.scale=1, var.scale=1, var.axes=T, varname.size = 2, varname.adjust = 2, )+ theme_bw()+ theme( legend.direction = 'horizontal', legend.position = 'top', legend.text = element_text(size = 10), legend.title = element_text(size = 10) ) ggsave('pca_gene.pdf',width=5,height=5,family="GB1") # PCA TYPE fig.A2 coln = colnames(ct) rown = rownames(ct) ct=t(ct) #Transpose colnames(ct)=rown rownames(ct)=coln ct <- ct[, colSums(ct != 0) > 0] ct.pca <- prcomp(ct,scale.=TRUE) ggbiplot( ct.pca, choices=c(1,2), obs.scale=1, var.scale=1, var.axes=F, labels=c(rownames(ct)), groups=c("GM","GM","GM","H1","H1","H1","H1") , ellipse = TRUE, ellipse.prob = 0.95, )+ scale_color_manual(values=c('#70abd8','#fb9b8e'))+ theme_bw()+ theme( legend.direction = 'horizontal', legend.position = 'top', legend.text = element_text(size = 10), legend.title = element_text(size = 10) ) ggsave('pca_type.pdf',width=5,height=5,family="GB1") -
画出四组差异基因的Venn图。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22# Venn Graph fig.B if (!require(VennDiagram)) BiocManager::install("VennDiagram") library(VennDiagram) gene.list = list(sig.deseq,sig.limma,sig.edgeR,sig.sam) venn1 <- venn.diagram( gene.list, category.names = c("deseq","limma","edgeR","sam"), alpha = 0.5, cex = 2, cat.cex = 2, cat.fontface = "bold", lty = 2, cat.pos = 0, filename = NULL, resolution = 300, fill = c("red", "blue", "green", "yellow"), compression = "lzw" ) pdf("venn.pdf",width = 10,height = 10) grid.draw(venn1) dev.off() -
对差异基因绘制热图
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34# Pheat map fig.C1 if (!require(pheatmap)) install.packages("pheatmap") library(pheatmap) save_pheatmap_pdf <- function(x, filename, width=7, height=7) { stopifnot(!missing(x)) stopifnot(!missing(filename)) pdf(filename, width=width, height=height) grid::grid.newpage() grid::grid.draw(x$gtable) dev.off() } common_diff_genes <- Reduce(intersect, list(sig.deseq,sig.edgeR,sig.limma,sig.sam)) ct <- count.table[rownames(count.table) %in% common_diff_genes,] ct = t(scale(t(ct))) annotation_col <- data.frame( celltype = c("GM","GM","GM","H1","H1","H1","H1") ) rownames(annotation_col) <- colnames(ct) pm <- pheatmap( ct, fontsize = 10, fontsize_row = 2, fontsize_col = 2, display_numbers = F, annotation_col = annotation_col, color = colorRampPalette(colors = c("blue","white","red"))(100) ) save_pheatmap_pdf(pm, "C:\\Users\\admin\\Desktop\\Rscripts\\pheatmap.pdf",6,12) # Pheat map fig.C2 pdf("hm2.pdf",width=5,height=5) heatmap(cor(ct),cexCol=0.75,cexRow=0.75) dev.off() -
绘制火山图、可视化差异基因的log2FC和P值。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24expr_matrix <- count.table[rownames(count.table) %in% common_diff_genes,] GM_samples <- 1:3 # 前三列为 GM H1_samples <- 4:7 # 后四列为 H1 sig2 <- sig[rownames(sig) %in% common_diff_genes,] Gene_table = data.frame(rownames(sig2),sig2$logFC,sig2$adj.P.Val) rownames(Gene_table) = Gene_table$rownames.sig2. Gene_table = Gene_table[,-1] colnames(Gene_table) <- c("log2FC","pvalue") change = as.factor(ifelse(Gene_table$pvalue < 0.05 & abs(Gene_table$log2FC) > 1, ifelse(Gene_table$log2FC > 1 ,'Up','Down'),'No change')) pdf("vol.pdf",width=5,height=5) ggplot(Gene_table,aes(log2FC, -log10(pvalue)))+ geom_hline(yintercept = -log10(0.05), linetype = "dashed", color = "#999999")+ geom_vline(xintercept = c(-1,1), linetype = "dashed", color = "#999999")+ geom_point(aes(color = change), size = 0.2, alpha = 0.5) + theme_bw(base_size = 12)+ ggsci::scale_color_jama() + theme(panel.grid = element_blank(), legend.position = 'right') dev.off()
图片